Patients Come First.
prod:prod_dcx-login


A Passion for Better Patient Care
At Cepheid, we know timing is critical to delivering better patient care. Our vision is simple: we believe in order to make a real difference, clinicians need answers now.
With our fast, accurate and easy-to-use molecular testing solutions, we are enabling people like you, working in healthcare facilities of any size, to get answers when and where you need them. With the power of Cepheid’s solutions, you can make the best clinical decisions to positively impact patient outcomes.
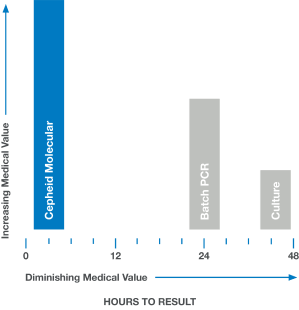
Medical Value:
Accelerating Time to Result
In medicine, time is often the enemy. Rapid and accurate answers are often imperative. When results are delayed, as with culture or batch-based PCR testing, medical value diminishes and clinical decisions are less likely to have a positive impact.

Video
Catawba Valley Medical Center Leverages the GeneXpert® System
Catawba Valley Medical Center leverages Cepheid’s GeneXpert® System to improve patient care and prevent infections.
Video
Catawba Valley Medical Center Leverages the GeneXpert® System
Catawba Valley Medical Center leverages Cepheid’s GeneXpert® System to improve patient care and prevent infections.
Making a Positive Impact, Together